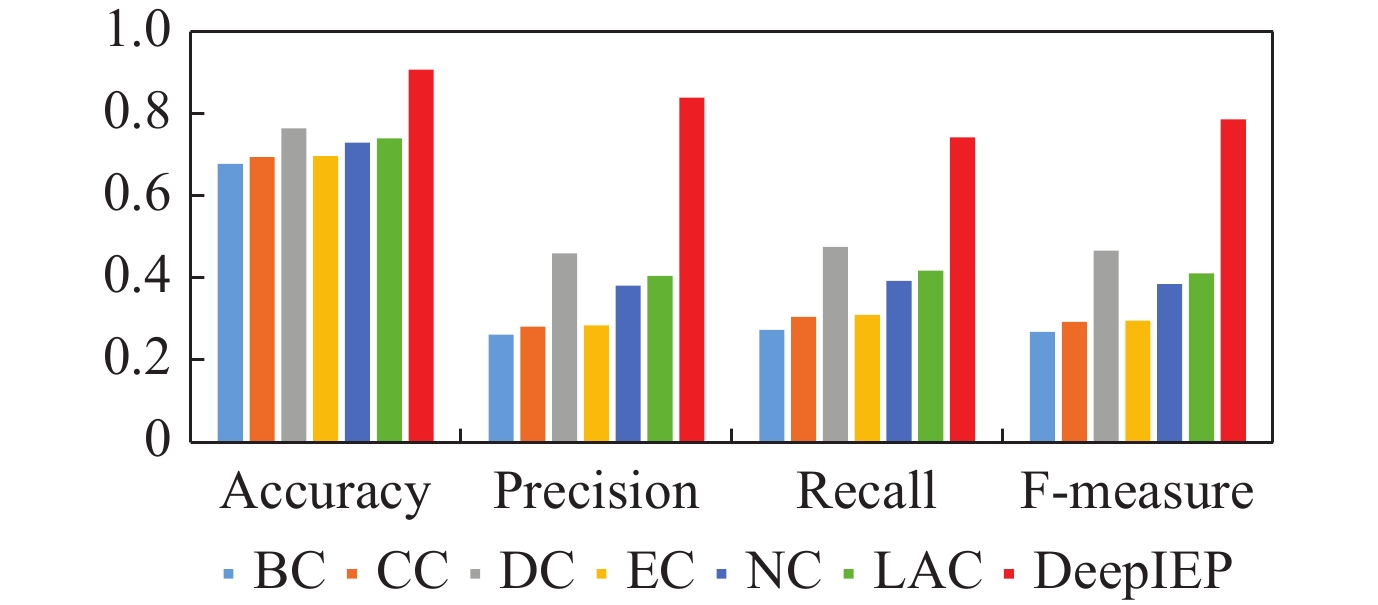
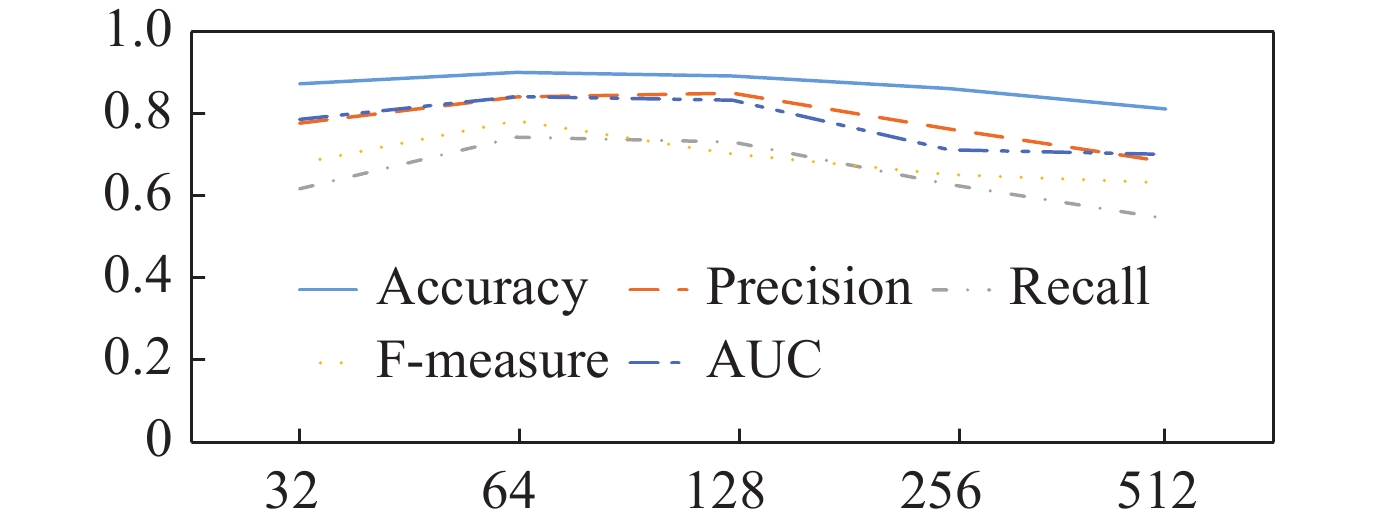
| Citation: | LI Weihua, LIU Wenyang, GUO Yanbu, et al., “Deep Contextual Representation Learning for Identifying Essential Proteins via Integrating Multisource Protein Features,” Chinese Journal of Electronics, vol. 32, no. 4, pp. 868-881, 2023, doi: 10.23919/cje.2022.00.053

|
| [1] |
W. He, L. Zhang, O. D. Villarreal, et al., “De novo identification of essential protein domains from CRISPR-Cas9 tiling-sgRNA knockout screens,” Nature Communications, vol.10, article no.articleno.4541, 2019. doi: 10.1038/s41467-019-12489-8
|
| [2] |
P. Y. Zhang, M. T. Zhang, H. Liu, et al., “Prediction of protein subcellular localization based on microscopic images via multi-task multi-instance learning,” Chinese Journal of Electronics, vol.31, no.5, pp.888–896, 2022. doi: 10.1049/cje.2020.00.330
|
| [3] |
X. Q. Yang, X. J. Lei, and J. Zhao, “Essential protein prediction based on shuffled frog-leaping algorithm,” Chinese Journal of Electronics, vol.30, no.4, pp.704–711, 2021. doi: 10.1049/cje.2021.05.012
|
| [4] |
M. R. Fan, M. Li, Z. F. Liu, et al., “Crystal structures of the PsbS protein essential for photoprotection in plants,” Nature Structural & Molecular Biology, vol.22, no.9, pp.729–735, 2015. doi: 10.1038/nsmb.3068
|
| [5] |
M. Li, R. Q. Zheng, H. H. Zhang, et al., “Effective identification of essential proteins based on priori knowledge, network topology and gene expressions,” Methods, vol.67, no.3, pp.325–333, 2014. doi: 10.1016/j.ymeth.2014.02.016
|
| [6] |
X. Y. Li, W. K. Li, M. Zeng, et al., “Network-based methods for predicting essential genes or proteins: A survey,” Briefings in Bioinformatics, vol.21, no.2, pp.566–583, 2020. doi: 10.1093/bib/bbz017
|
| [7] |
L. M. Cullen and G. M. Arndt, “Genome-wide screening for gene function using RNAi in mammalian cells,” Immunology & Cell Biology, vol.83, no.3, pp.217–223, 2005. doi: 10.1111/j.1440-1711.2005.01332.x
|
| [8] |
T. Roemer, B. Jiang, J. Davison, et al., “Large-scale essential gene identification in Candida albicans and applications to antifungal drug discovery,” Molecular Microbiology, vol.50, no.1, pp.167–181, 2003. doi: 10.1046/j.1365-2958.2003.03697.x
|
| [9] |
H. Jeong, S. P. Mason, A. L. Barabási, et al., “Lethality and centrality in protein networks,” Nature, vol.411, no.6833, pp.41–42, 2001. doi: 10.1038/35075138
|
| [10] |
M. W. Hahn and A. D. Kern, “Comparative genomics of centrality and essentiality in three eukaryotic protein-interaction networks,” Molecular Biology and Evolution, vol.22, no.4, pp.803–806, 2005. doi: 10.1093/molbev/msi072
|
| [11] |
M. P. Joy, A. Brock, D. E. Ingber, et al., “High-betweenness proteins in the yeast protein interaction network,” Journal of Biomedicine and Biotechnology, vol.2005, no.2, article no.594674, 2005. doi: 10.1155/JBB.2005.96
|
| [12] |
S. Wuchty and P. F. Stadler, “Centers of complex networks,” Journal of Theoretical Biology, vol.223, no.1, pp.45–53, 2003. doi: 10.1016/S0022-5193(03)00071-7
|
| [13] |
E. Estrada and J. A. Rodriguez-Velazquez, “Subgraph centrality in complex networks,” Physical Review E, vol.71, no.5, article no.056103, 2005. doi: 10.1103/PhysRevE.71.056103
|
| [14] |
P. Bonacich, “Power and centrality: A family of measures,” American Journal of Sociology, vol.92, no.5, pp.1170–1182, 1987. doi: 10.1086/228631
|
| [15] |
K. Stephenson and M. Zelen, “Rethinking centrality: Methods and examples,” Social Networks, vol.11, no.1, pp.1–37, 1989. doi: 10.1016/0378-8733(89)90016-6
|
| [16] |
M. Zeng, M. Li, Z. H. Fei, et al., “A deep learning framework for identifying essential proteins by integrating multiple types of biological information,” IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol.18, no.1, pp.296–305, 2021. doi: 10.1109/TCBB.2019.2897679
|
| [17] |
E. Nasiri, K. Berahmand, M. Rostami, et al., “A novel link prediction algorithm for protein-protein interaction networks by attributed graph embedding,” Computers in Biology and Medicine, vol.137, article no.104772, 2021. doi: 10.1016/j.compbiomed.2021.104772
|
| [18] |
G. S. Li, M. Li, J. X. Wang, J. et al., “Predicting essential proteins based on subcellular localization, orthology and PPI networks,” BMC Bioinformatics, vol.17, no.S8, article no.279, 2016. doi: 10.1186/s12859-016-1115-5
|
| [19] |
A. M. Gustafson, E. S. Snitkin, S. C. J. Parker, et al., “Towards the identification of essential genes using targeted genome sequencing and comparative analysis,” BMC Genomics, vol.7, article no.265, 2006. doi: 10.1186/1471-2164-7-265
|
| [20] |
J. C. Zhong, J. X. Wang, W. Peng, et al., “Prediction of essential proteins based on gene expression programming,” BMC Genomics, vol.14, no.S4, article no.S7, 2013. doi: 10.1186/1471-2164-14-S4-S7
|
| [21] |
X. Y. Zhu, Y. C. Zhu, Y. H. Tan, et al., “An iterative method for predicting essential proteins based on multifeature fusion and linear neighborhood similarity,” Frontiers in Aging Neuroscience, vol.13, article no.799500, 2022. doi: 10.3389/FNAGI.2021.799500
|
| [22] |
L. Wang, J. X. Peng, L. N. Kuang, et al., “Identification of essential proteins based on local random walk and adaptive multi-view multi-label learning,” IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol.19, no.6, pp.3507–3516, 2022. doi: 10.1109/TCBB.2021.3128638
|
| [23] |
Y. C. Hwang, C. C. Lin, J. Y. Chang, et al., “Predicting essential genes based on network and sequence analysis,” Molecular BioSystems, vol.5, no.12, pp.1672–1678, 2009. doi: 10.1039/b900611g
|
| [24] |
J. Y. Deng, L. Deng, S. C. Su, et al., “Investigating the predictability of essential genes across distantly related organisms using an integrative approach,” Nucleic Acids Research, vol.39, no.3, pp.795–807, 2011. doi: 10.1093/nar/gkq784
|
| [25] |
A. K. Payra and A. Ghosh, “Identifying essential proteins using modified-monkey algorithm (MMA),” Computational Biology and Chemistry, vol.88, article no.107324, 2020. doi: 10.1016/j.compbiolchem.2020.107324
|
| [26] |
M. Zeng, M. Li, F. X. Wu, et al., “DeepEP: A deep learning framework for identifying essential proteins,” BMC Bioinformatics, vol.20, no.S16, article no.506, 2019. doi: 10.1186/s12859-019-3076-y
|
| [27] |
X. Zhang, W. X. Xiao, and W. J. Xiao, “DeepHE: Accurately predicting human essential genes based on deep learning,” PLoS Computational Biology, vol.16, no.9, article no.e1008229, 2020. doi: 10.1371/journal.pcbi.1008229
|
| [28] |
M. H. Chen, C. J. T. Ju, G. Y. Zhou, et al., “Multifaceted protein-protein interaction prediction based on Siamese residual RCNN,” Bioinformatics, vol.35, no.14, pp.i305–i314, 2019. doi: 10.1093/bioinformatics/btz328
|
| [29] |
Y. B. Guo, B. Y. Wang, W. H. Li, et al., “Protein secondary structure prediction improved by recurrent neural networks integrated with two-dimensional convolutional neural networks,” Journal of Bioinformatics and Computational Biology, vol.16, no.5, article no.1850021, 2018. doi: 10.1142/S021972001850021X
|
| [30] |
Y. B. Guo, D. M. Zhou, R. C. Nie, et al., “DeepANF: A deep attentive neural framework with distributed representation for chromatin accessibility prediction,” Neurocomputing, vol.379, pp.305–318, 2020. doi: 10.1016/j.neucom.2019.10.091
|
| [31] |
M. Ghandi, D. Lee, M. Mohammad-Noori, et al., “Enhanced regulatory sequence prediction using gapped k-mer features,” PLoS Computational Biology, vol.10, no.7, article no.e1003711, 2014. doi: 10.1371/journal.pcbi.1003711
|
| [32] |
A. Grover and J. Leskovec, “node2vec: Scalable feature learning for networks,” in Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, pp.855–864, 2016.
|
| [33] |
H. C. Gao and H. Huang, “Deep attributed network embedding”, in Proceedings of the Twenty-Seventh International Joint Conference on Artificial Intelligence, Stockholm, Sweden, pp.3364–3370, 2018.
|
| [34] |
X. W. Tang, J. X. Wang, J. C. Zhong, et al., “Predicting essential proteins based on weighted degree centrality,” IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol.11, no.2, pp.407–418, 2014. doi: 10.1109/TCBB.2013.2295318
|
| [35] |
M. Li, H. H. Zhang, J. X. Wang, et al., “A new essential protein discovery method based on the integration of protein-protein interaction and gene expression data,” BMC Systems Biology, vol.6, article no.15, 2012. doi: 10.1186/1752-0509-6-15
|
| [36] |
K. Plaimas, R. Eils, and R. König, “Identifying essential genes in bacterial metabolic networks with machine learning methods,” BMC Systems Biology, vol.4, article no.56, 2010. doi: 10.1186/1752-0509-4-56
|
| [37] |
D. S. Huang and C. H. Zheng, “Independent component analysis-based penalized discriminant method for tumor classification using gene expression data,” Bioinformatics, vol.22, no.15, pp.1855–1862, 2006. doi: 10.1093/bioinformatics/btl190
|
| [38] |
R. Fakoor, F. Ladhak, A. Nazi, et al., “Using deep learning to enhance cancer diagnosis and classification,” in Proceedings of the 30th International Conference on Machine Learning, Atlanta, GA, USA, pp.3937–3949, 2013.
|
| [39] |
Y. B. Guo, W. H. Li, B. Y. Wang, et al., “DeepACLSTM: Deep asymmetric convolutional long short-term memory neural models for protein secondary structure prediction,” BMC Bioinformatics, vol.20, article no.341, 2009. doi: 10.1186/s12859-019-2940-0
|
| [40] |
N. Altwaijry and I. Al-Turaiki, “Arabic handwriting recognition system using convolutional neural network,” Neural Computing and Applications, vol.33, no.7, pp.2249–2261, 2021. doi: 10.1007/s00521-020-05070-8
|
| [41] |
Z. Cheng, L. Liu, G. L. Lin, et al., “ReHiC: Enhancing Hi-C data resolution via residual convolutional network,” Journal of Bioinformatics and Computational Biology, vol.19, no.2, article no.2150001, 2021. doi: 10.1142/S0219720021500013
|
| [42] |
M. Alkhodari and L. Fraiwan, “Convolutional and recurrent neural networks for the detection of valvular heart diseases in phonocardiogram recordings,” Computer Methods and Programs in Biomedicine, vol.200, article no.105940, 2021. doi: 10.1016/j.cmpb.2021.105940
|
| [43] |
J. B. Wang, “Automated detection of premature ventricular contraction based on the improved gated recurrent unit network,” Computer Methods and Programs in Biomedicine, vol.208, article no.106284, 2021. doi: 10.1016/J.CMPB.2021.106284
|
| [44] |
The PLOS Computational Biology Staff, “Correction: Enhanced regulatory sequence prediction using gapped k-mer features,” PLoS Computational Biology, vol.10, no.7, article no.e1004035, 2014. doi: 10.1371/journal.pcbi.1004035
|
| [45] |
V. Nair and G. E. Hinton, “Rectified linear units improve restricted boltzmann machines,” in Proceedings of the 27th International Conference on Machine Learning, Haifa, Israel, pp.807–814, 2010.
|
| [46] |
D. P. Kingma and J. Ba, “Adam: A method for stochastic optimization,” in Proceedings of the 3rd International Conference on Learning Representations, San Diego, CA, USA, 2015.
|
| [47] |
C. Stark, B. J. Breitkreutz, T. Reguly, et al., “BioGRID: A general repository for interaction datasets,” Nucleic Acids Research, vol.34, no.S1, pp.D535–D539, 2006. doi: 10.1093/nar/gkj109
|
| [48] |
I. Xenarios, L. Salwinski, X. J. Duan, et al., “DIP, the Database of Interacting Proteins: A research tool for studying cellular networks of protein interactions,” Nucleic Acids Research, vol.30, no.1, pp.303–305, 2002. doi: 10.1093/nar/30.1.303
|
| [49] |
B. P. Tu, A. Kudlicki, M. Rowicka, et al., “Logic of the yeast metabolic cycle: Temporal compartmentalization of cellular processes,” Science, vol.310, no.5751, pp.1152–1158, 2005. doi: 10.1126/science.1120499
|
| [50] |
J. X. Wang, M. Li, H. Wang, et al., “Identification of essential proteins based on edge clustering coefficient,” IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol.9, no.4, pp.1070–1080, 2012. doi: 10.1109/TCBB.2011.147
|
| [51] |
M. Li, J. X. Wang, X. Chen, et al., “A local average connectivity-based method for identifying essential proteins from the network level,” Computational Biology and Chemistry, vol.35, no.3, pp.143–150, 2011. doi: 10.1016/j.compbiolchem.2011.04.002
|
| [52] |
Z. Zhang, H. X. Yang, J. J. Bu, et al., “ANRL: attributed network representation learning via deep neural networks”, in Proceedings of the Twenty Seventh International Joint Conference on Artificial Intelligence, Stockholm, Sweden, pp.3155–3161, 2018.
|
| [53] |
H. Öztürk, A. Özgür, and E. Ozkirimli, “DeepDTA: Deep drug-target binding affinity prediction,” Bioinformatics, vol.34, no.17, pp.i821–i829, 2018. doi: 10.1093/bioinformatics/bty593
|
| [54] |
L. Wang, H. Li, Y. Q. Wang, et al., “MDADP: A webserver integrating database and prediction tools for microbe-disease associations,” IEEE Journal of Biomedical and Health Informatics, vol.26, no.7, pp.3427–3434, 2022. doi: 10.1109/JBHI.2022.3156166
|
| [55] |
P. Y. Ping, L. Wang, L. N. Kuang, et al., “A novel method for LncRNA-Disease association prediction based on an lncRNA-Disease association network,” IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol.16, no.2, pp.688–693, 2019. doi: 10.1109/TCBB.2018.2827373
|
Figures(5) / Tables(6)

CJE QR CODE

CIE QR CODE
Copyright © Editorial Office of Chinese Journal of Electronics | 京ICP备12041980号-1 |
Supported by:
Beijing Renhe Information Technology Co. Ltd





 DownLoad:
DownLoad: